W. Szczepankiewicz, „Ogoreny”, Teneochem.ovh, 2024.01.29.
Proszę mi wybaczyć tę nazwę, ale pierwsze moje skojarzenie padło na ogórki. Nic na to nie poradzę. Wobec ogromnej liczby innych struktur. Muszę nazywać jakoś swoje pomysły chemiczne. Muszą to być nazwy unikalne i niekoniecznie bardzo sensowne. Tak się stało w wypadku tych struktur. Oczywiście można dodać ich nazwy systematyczne, co zapewne uczynię bez ostrzeżenia.
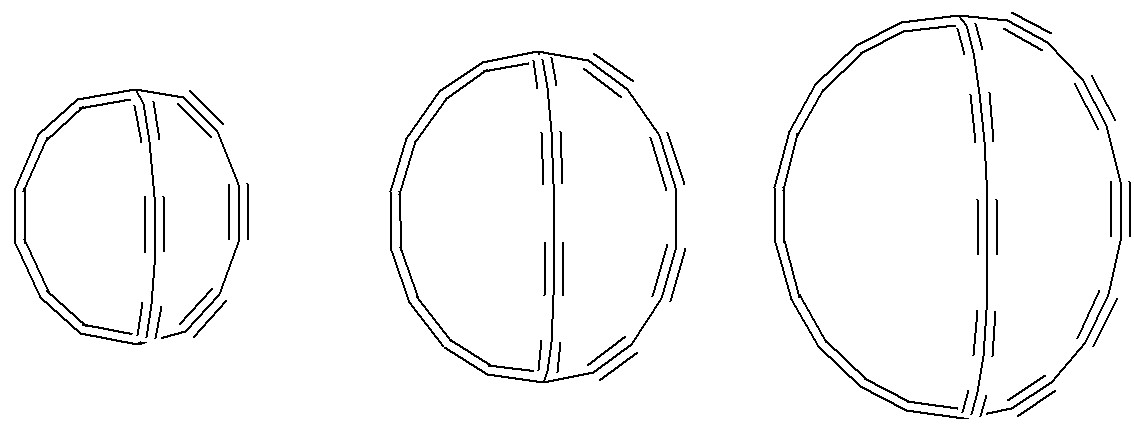
Pierwszym skonstruowanym związkiem był układ bicyklo[6.6.6]ikoza-2,4,6,9,11,13,15,17,19-nonayn. Wszystkie poniżej struktury zostały zoptymalizowane na poziomie DFT (Orca4.2.1/B3LYP/def2-SVP/hess-plus):
Kąty walencyjne H-C-C wynoszą 133.5° natomiast kąty C-C-C z wierzchołkiem dla fragmentu C-H wynoszą 105.1°. Oznacza to pewne odkształcenie od struktury tetraedrycznej wywołane kompromisem geometrycznym pomiędzy napięciem powstałym w nieliniowych fragmentach acetylenowych i idealnym ostrosłupem.
W pokazanym poniżej wydłużonym analogu, czyli bicyklo[8.8.8]heksakoza-2,4,6,8,11,13,15,17,19,21,23,25-dodekaynie, kąty te wynoszą odpowiednio 112.7° oraz 106.0°. Sugeruje to zmniejszenie naprężeń, ale nie ich całkowitą likwidację. Nie badałem bardziej wydłużonych łańcuchów ogorenów, ale myślę, że ta tendencja relaksacji struktury powinna być zachowana.
Poprzez usunięcie atomów wodoru z powyżej przedstawionych związków (i jednego skonstruowanego dodatkowo) i reorganizację wiązań wielokrotnych zostały uzyskane trzy odmiany alotropowe węgla:
Co ciekawe, moduł nazewniczy programu ChemSketch2020 potrafił nadać nazwy również przedstawionym powyżej alotropom. Model C20 nazwał bicyklo[6.6.6]ikoza-1,2,3,4,5,6,7-heptaeno-9,11,13,15,17,19-hexaynem. To jest ciekawe, gdyż przedstawione alotropy nie powinny być traktowane jak związki chemiczne. Tym niemniej w ten sposób została zachowana jednolitość nazewnictwa.
Ostatnim etapem prac nad strukturami alotropowymi było sprawdzenie, czy mogą tworzyć się trwałe połączenia pomiędzy bicyklami. W celu sprawdzenia skonstruowałem układ składający się z dwóch fragmentów oddalonych od siebie o 1.58 Å. Obliczenia zostały przeprowadzone na poziomie DFT (Orca4.2.1/revPBE/def2-TZVP):
Należy zwrócić uwagę na to, że nie jest to ścieżka reakcji ale postęp optymalizacji energii układu. Na tym etapie można stwierdzić, że cząsteczka połączona wiązaniem pojedynczym rozpada się na dwie odrębne. Być może jednak odległość początkowa pomiędzy atomami węgla, które tworzą wiązanie jest zbyt duża. To należy sprawdzić.