Historia tej grupy związków zaczęła się w kwietniu 2023 r. Wtedy skonstruowałem cząsteczkę, w której centrum znajduje się fragment cyklobutenowy (cząsteczka A na rysunku poniżej po optymalizacji za pomocą MM/ ChemSketch 2023):
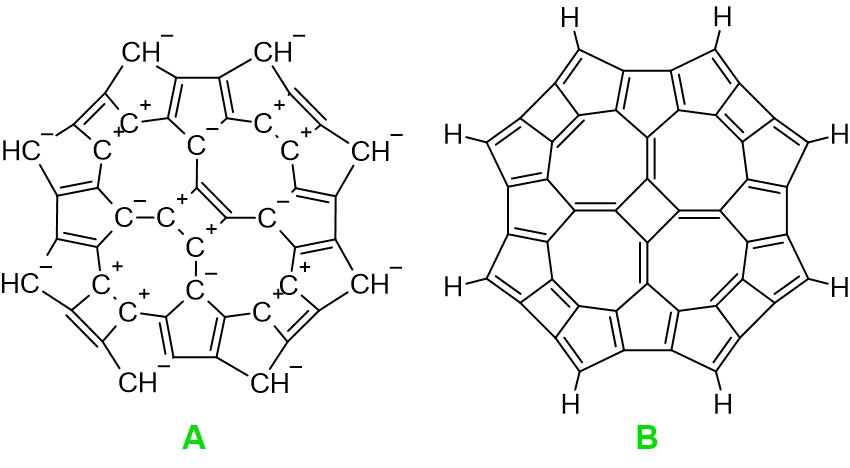
Wtedy starałem się tak dobierać fragmenty w cząsteczce A, aby wykazywały one charakter aromatyczny. Dlatego pojawiają się aniony cyklopentadienylowe oraz dikationy cyklobutenowe. Ta metoda konstrukcji powoduje, że cząsteczka ma ładunek dwuujemny (C48H8-2). Po pewnym czasie uświadomiłem sobie, że ładunki można kompensować tworząc po prostu wiązania podwójne (cząsteczka B). Cząsteczkę B poddałem optymalizacji (Orca 4.2.1/ PM3/ hess-plus):
Powyższa animacja pokazuje, że o ile MM przewiduje, że cząsteczka jest płaska, o tyle na poziomie półmepirycznym optymalizacja struktury wyjściowej (którą jest struktura zoptymalizowana za pomocą MM) powoduje przejście do kształtu podobnego do koszyczka.
Połączyłem ze sobą takie dwa „koszyczki” usuwając wcześniej atomy wodoru i łacząc zwolnione miejsca za pomocą wiązań C-C. Zabieg ten pozwolił na stworzenie alotropu węgla C96 (Orca 4.2.1/ PM3/ hess-plus):
Sugerując się strukturą A zoptymalizowałem również tetraanion na poziomie półempirycznym (parametry obliczeń jak wyżej). Jego geometria jest podobna do alotropu nienaładowanego, choć oczywiście różni się w szczegółach.
Zadałem sobie pytanie, jak będzie wyglądać cząsteczka, gdyby centralny pierścień czteroczłonowy zastąpić pierścieniem pięcioczłonowym. Przeorganizowanie struktury dało cząsteczkę o wzorze C55H16 (oznaczę go jako C). Nieparzysta liczna atomów węgla sugeruje, że układ wiązań podwójnych w cząsteczce nie może być w pełni sprzężony:
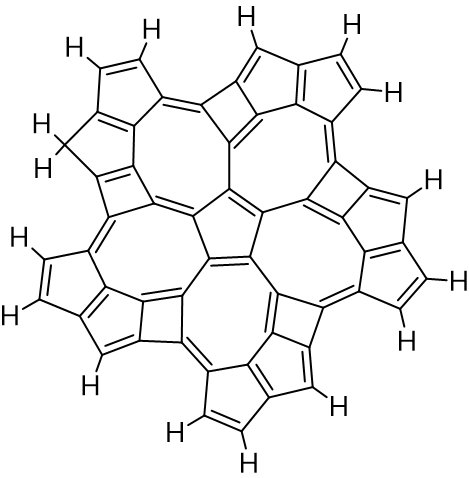
Optymalna geometria obliczona na poziomie półempirycznym (parametry obliczeń jak wyżej) ma charakter spłaszczonego siodła:
Podobnie jak poprzednio skonstruowałem model alotropu przez usunięcie atomów wodoru z dwóch cząsteczek w powyższej strukturze i połączenie tak zwolnionych wiązań za pomocą wiązań C-C. W wypadku grup CH2 zastąpiło je wiązanie C=C. Powstały w ten sposób alotrop nie jest pofałdowany (parametry obliczeń jak wyżej):
Zoptymalizowałem również na poziomie półempirycznym (MOPAC 22.06/ PM7/ hessplus) dianion tego alotropu.
Skonstruowałem jeszcze jeden alotrop. Dwie cząsteczki C pozbawione atomów wodoru połączyłem ze sobą w dość ryzykowny sposób. Dwa atomy węgla sp3 połączyłem wiązaniami pojedynczymi z atomami węgla opisanymi za pomocą hybrydyzacji sp2. Ryzyko polegało na tym, że powinienem do tetraedrycznych atomów węgla dobudować po jednym atomie wodoru. Nie uczyniłem tego licząc na to, że i tak utworzy się sprzężony układ wiązań podwójnych (Orca 4.2.1/ PM3/ hess-plus:.
Rzeczywiście struktura zoptymalizowanej cząsteczki posiada całkowicie sprzężony układ wiązań podwójnych. Nie powinno być to dziwne, gdyż zawiera parzystą liczbę atomów węgla (110 atomów).