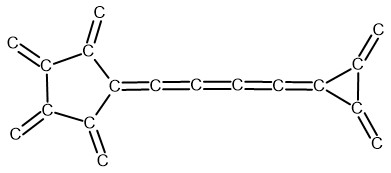
Nazwa jest związana ze skojarzeniem kształtu tych alotropów węgla z kształtem piłki do koszykówki. Nazwy i ich dodatkowe oznaczeni nie mają tu jednak większego znaczenia. Pomagają mi w pamiętaniu grup struktur, które skonstruowałem. Pierwszy alotrop skonstruowany jest z pierścieni cyklopentanowych, cyklopropanowych oraz łańcuchów butatrienowych. Poniżej zamieszczony rysunek fragnetu pilkenów pokazuje ogólny zamysł budowy tych cząsteczek:
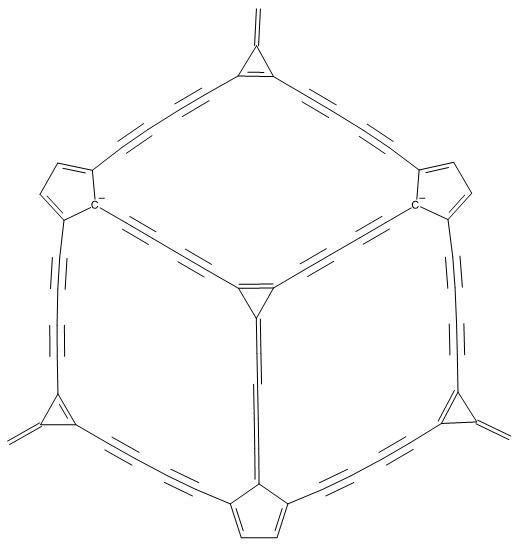
Rozbudowa powyższego fragmentu polegała na dodaniu do wolnych wartościowości czterowęglowych nienasyconych łańcuchów oraz pierścieni trój praz pięcioczłonowych w rozmaitych konfiguracjach. Te konfiguracje miały być tak skonstruowane aby finalnie uzyskać kształty kuliste i owalne mające charakter alotropów węgla. Badałem również inne konfiguracje połączeń pierścieni (patrz niżej pilkenA).
Rysunek pokazany poniżej pokazuje zoptymalizowaną cząsteczkę pilkenuA (Orca4.2.1/B3LYP/def2-SVP/hess-plus). Jak widać 3DMol dość swobodnie interpretuje krotności wiązań:
Należy przyjąć, że pierścienie połączone są serią sprzężonych wiązań podwójnych. Dopiero wówczas wzór Lewisa powyższej struktury przyjmuje prawidłową postać. Tę poprawną sytuację elektronową pokazuje poniżej zamieszczony rysunek fragmentu pilkenu (ChemSketch2022):
PilkenB zorganizowany jest inaczej. Można tu wyróżnić dwie części bardziej płaskie połączone wygiętymi łańcuchami trienowymi (Orca4.2.1/HF/def2-SVP/hess-plus):
W tym pilkenie łańcuchy trienowe łączą tego samego rozmiaru fragmenty cykliczne. W odróżnieniu do tego w pilkenieA, wyselekcjonowanie analogicznych „płaszczyzn” jest trudne (nie są płaskie a raczej mają kształt czasz). Ważne jednak, że bardziej wypukłe analogi płaszczyzn są ze sobą połączone inaczej. Tutaj pierścienie pięcioczłonowe jednej części łączą się z pierścieniami trójczłonowymi drugiej i odwrotnie.
PilkenC został skonstruowany z sześciu pierścieni pięcioczłonowych i ośmiu pierścieni trójczłonowych. Niestety po udanej optymalizacji pojawiły się problemy zarówno na poziomie półempirycznym (MOPAC22/PM7/precise) jaki i HF (baza 6-31G). Pojawienie się wielu częstości ujemnych i charakter ich „drgań” wskazuje, że źródłem tych problemów są naprężenia wynikające z obecności pierścieni trójczłonowych. Mimo tych problemów pokazuję poniżej optymalną ale niezrelaksowaną strukturę tego pilkenu:
W pilkenieE3 cztery pierścienie trójczłonowe „rozszczepiłem” pozostawiając jeden z atomów jako węzeł łańcuchów. Ten zabieg spowodował zrelaksowanie naprężeń i obliczenia częstości na poziomie półempirycznym wypadły pomyślnie (MOPAC22/PM7/hess-plus):
Sytuacja niecałkowitej relaksacji zauważalna jest również w izomerze pilkenuE. Ten izomer jest tu oznaczone jako pilkenD. Jest to izomer pilkenuE3 z ośmioma pierścieniami trójczłonowymi:
Próba relaksacji na poziomie DFT (choć z ograniczoną bazą funkcyjną – Orca4.2.1/HF/6-31G) dała jednak jedną częstość ujemną (-7.78 cm-1). Poniższa animacja pokazuje zachowanie się tej częstości (wizualizacja program Avogadro 1.99/OSD/CapCut):
Film wskazuje na kierunek zmian geometrycznych. Drganie „dąży” do rozerwania jednego z wiązań tworzących pierścień trójczłonowy (niestety program Avogadro pokazuje pięciowiązalne atomy węgla, cóż…). Wydaje się, że ręczna likwidacja tego pierścienia i utworzenie węzła z atomu węgla, może usunąć ujemną częstość (podobnie jak w pilkenieE3). Tego nie badałem i rozwiązanie tego problemu pozostaje zagadką (marzec 2024).
Ostatnia cząsteczka policzona w tej serii to pilkenF. Cząsteczka tego alotropu składa się z 360 atomów węgla. Liczba atomów jest zbyt duża, aby dało się obliczyć na moim komputerze (z 16 rdzeniami) częstości na poziomie wyższym niż półempiryczny w akceptowalnym czasie. Początkowo miałem trudności nawet ze wstępną optymalizacją tej struktury. Zarówno MOPAC22 (PM7/EF) jak i Orca4.2.1 (PM3/standardowe parametry optymalizacji) wykazywały problemy z zakończeniem szukania minimum energii potencjalnej. Dopiero metoda AM1 ze standardowymi parametrami optymalizacji (ale ze wstępnie „podoptymalizowanych” współrzędnych z obliczeń PM3 – Orca4.2.1/AM1/SOSCF/hess-plus) pozwoliła na uzyskanie optymalnej geometrii jak i wszystkich częstości dodatnich w hessianie:
Jak się okazało, w wypadku programu Orca, problem wynikał z zastosowania procedury optymalizacyjnej SOSCF. Wyjściowa struktura była zbyt odległa od optimum, co powodowało załamywanie się optymalizacji. Dopiero zmiana procedury za pomocą komendy NOSOSCF spowodowała, że optymalizacja przebiegała bez zakłóceń, ale była bardzo wolna. Po 12 godzinach obliczeń przerwałem działanie programu i użyłem otrzymaną przybliżoną strukturę (w trybie NOSOSCF) w kolejnej optymalizacji (SOSCF). Tym razem wychodząc z lepiej zoptymalizowanej geometrii procedura nie ulegała zawieszeniu i dość szybko program osiągnął minimum energii i przeszedł do obliczeń częstości. Dzięki zmianie techniki obliczeń udało się również uzyskać zrelaksowaną cząsteczkę za pomocą metody PM3 (oznaczonej technicznie jako pilkenK).
Wydaje się też, że powyższe struktury mogą występować w postaci karbojonów z parzystą liczbą elektronów. Nie badałem jednak ich właściwości metodami obliczeniowymi. Ten problem czeka na rozpatrzenie (marzec 2024).