4. Wyniki obliczeń wybranych modeli
4.1. Uwaga wstępna
Należy solennie przyznać, że tylko niewielka liczba z narysowanych w pierwszej części struktur została poddana optymalizacji na poziomie półempirycznym i DFT. Tym niemniej obliczeniom poddałem związki zawierające różne liczby jednostek wyjściowych modeli. Wynikało to z mojego początkowego podejścia. Postaram się w skrótowy sposób opisać uzyskane wyniki. Do obliczeń wybrałem modele z największą liczbą wiązań podwójnych, to znaczy C oraz H. Sądziłem, że tutaj może być najwięcej niespodzianek strukturalnych, to znaczy takich, gdzie wyniki optymalizacji na poziomach półempirycznym i DFT mogą różnić się od struktur założonych na wstępnie i zoptymalizowanych za pomocą mechaniki molekularnej. Tak też się stało w kilku wypadkach.
4.2. Wyniki obliczeń dla modeli C
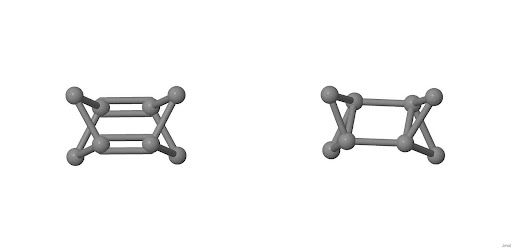
Cząsteczka C podstawowa, która w zamyśle miała być układem tricyklicznym z dwoma wiązaniami podwójnymi okazała się po obliczeniach na poziomie DFT związkiem z wiązaniami pojedynczymi, czyli pentacyklo[3.1.1.12,4.01,5.02,4]oktanem (rysunek 4.1.):
W następnych krokach rozbudowie i optymalizacji poddano model C po wstępnym uporządkowaniu geometrii wstępnych modeli za pomocą mechaniki molekularnej. Wielkość m przyjmuje wartości od 0 do 15 ze skokiem jeden. Należy pamiętać, że dla nieparzystych wartości m skrzydłowe grupy CH2 na obu końcach cząsteczki leżą na prostych prostopadłych (schemat 4.1. niestety nie oddaje tego w sposób dostatecznie klarowny):
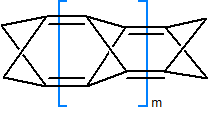
Struktura pozostałych modeli po optymalizacji na poziomie półempirycznym lub DFT zależy od wartości wskaźnika m. Wyniki te przedstawia tabela 4.1. Kolor niebieski tła komórki w tabeli oznacza, że wyjściowe cząsteczki zachowały ogólną strukturę substratu po optymalizacji na poziomie półempirycznym, co najwyżej ze zmianą położenia niektórych wiązań wewnętrznych, oraz że ich geometria została zweryfikowana na poziomie DFT. Kolor czerwony tła oznacza, że cząsteczki uległy przegrupowaniom w czasie obliczeń na poziomie półempirycznym. W tym wypadku nie prowadzono weryfikującej optymalizacji na poziomie DFT (chyba, że cząsteczka była zbyt ładna, aby to pominąć):
Tu powinna być tabela
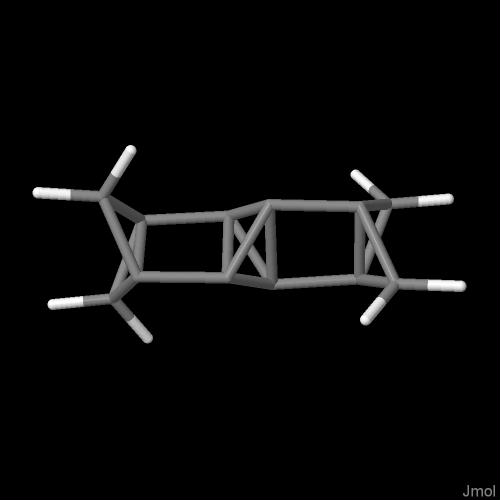
Dla wartości m = 1 skrzydłowe pierścienie przyjmują charakter bicyklo[1.1.0]butanów a środkowy fragment pochodzi od tricyklo[1.1.0.02,4]butanu (schemat 4.2.):
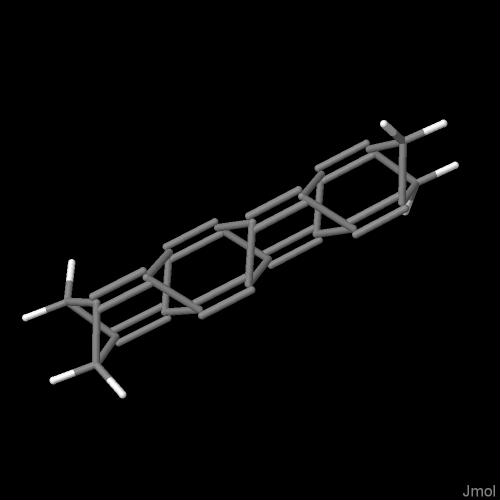
Dla m = 2 układ wiązań staje się bardziej skomplikowany. Wewnątrz cząsteczki pojawia się fragment cyklobutadienu, wyjściowa para wiązań podwójnych i fragmenty oligocykliczne. Nie należy jednak przywiązywać zbytniej wagi do sposobu wyświetlania wiązań wielokrotnych w tym szeregu cząsteczek. Różne programy wizualizacyjne robią to różnie. Rozkład wiązań wewnętrznych tego samego pliku wynikowego jest różnie interpretowany przez procedury graficzne, przykładowo programu jmol a inaczej przez Gabedit. Bez oporów można przyjąć, że wszystkie cykle poprzeczne składają się z bicyklo[1.1.0]butanów.
Jednak zasadniczy szkielet zostaje zachowany tylko dla części modeli (m = 0,1,2,4,6,10,13 i 14). Dla pozostałych optymalizacja na poziomie półempirycznym powoduje dramatyczne zmiany strukturalne. Tak się dzieje np. dla modelu C, gdzie m = 3.
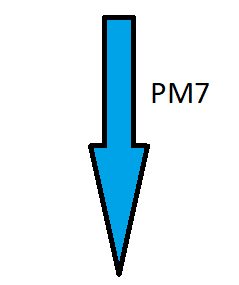
Obniżenie energii układu następuje poprzez utworzenie dwóch pierścieni oktadiynowych połączonych mostkiem tricyklo[1.1.0.02,4]butanu. Na skrzydłach znajdują się bicyklo[1.1.0]butany.
Podobnie zachowują się cząsteczki dla m = 5,7,8,9,11,12. Jednakże w tych wypadkach powstają związki o dość przypadkowym, jak się zdaje, ułożeniu cyklicznych elementów strukturalnych (scheme 4.3):
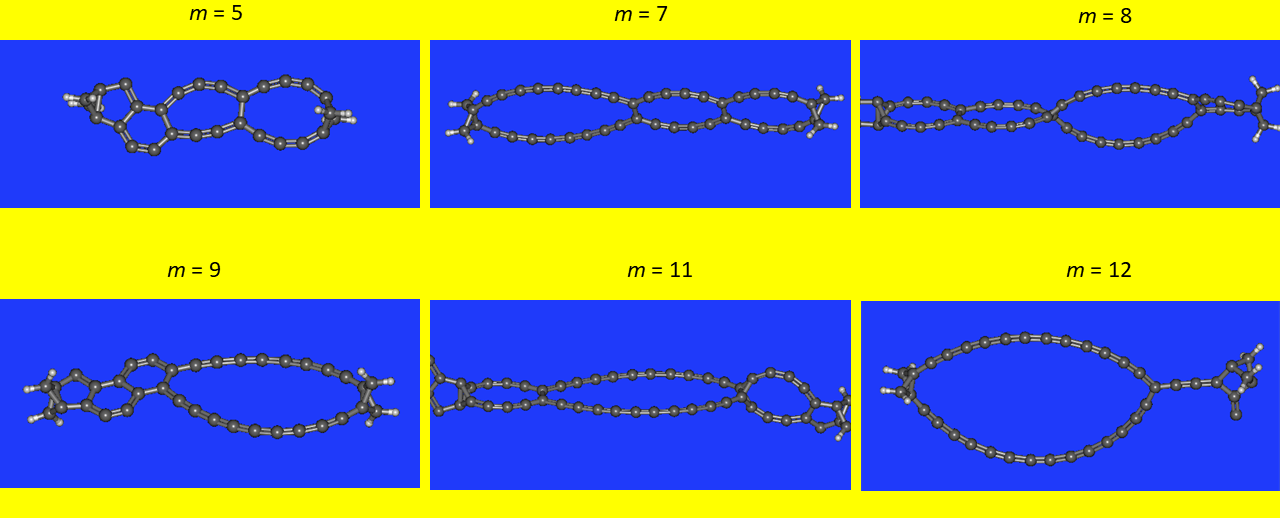
Cechą charakterystyczną wyżej pokazanych przykładów są długołańcuchowe fragmenty sprzężonych polienów. Podobne długołańcuchowe polieny sprzężone wykorzystałem w wielu następnych kreacjach.
Nie jest jasne, dlaczego dla niektórych związków wyjściowych ich początkowy szkielet jest zachowany w czasie optymalizacji, a w innych przypadkach następuje przegrupowanie. Być może wynik optymalizacji za pomocą metody PM7 zależy od dokładności przybliżenia geometrii przez mechanikę molekularną. Nie jest również wykluczone, że procedury optymalizacyjne są czułe na warunki początkowe i zachowują się podobnie do efektu motyla z teorii chaosu. Być może bariery energetyczne wokół lokalnych minimów części związków są niskie i procedury znajdują minima o niższej energii w innych przypadkach te bariery są na tyle wysokie, że oryginalny szkielet zostaje zachowany. Ta kwestia pozostaje otwarta.
4.3. Wyniki dla modeli H
Cząsteczka H (schemat 4.n dla m = 0) zawiera dwa pierścienie cyklobutadienowe połączone parą wiązań pojedynczych. Należy zwrócić uwagę na to, że obie pary skrzydłowych atomów wodoru leżą na tej samej prostej tak samo się dzieje dla każdej parzystej liczby pierścieni cyklobutadienowych (m = 0,2,4,6…14).
Model H (formalnie tricyklo[3.1.1.12,4]okta-1(7),2(8),3,5-tetraene) w zamyśle miał posiadać dwa pierścienie cyklobutadienowe połączone dwoma wiązaniami pojedynczymi w pozycjach 1,1’ oraz 3,3’. Jak wiadomo, układy antyaromatyczne są jednak nietrwałe. I rzeczywiście, optymalizacja modelu H na poziomie PM7 oraz DFT pokazuje, że związek ten ulega przegrupowaniu do dirodnika będącego formalnie (1p,4p)-bicyklo[4.1.1]octa-1,2,4,5-tetraene-7,8-diyl.
Optymalna odległość pomiędzy atomami węgla z niesparowanymi elektronami wynosi 2.22 Å. Nie tworzy się więc wiązanie pomiędzy tymi atomami i układ zdaje się mieć charakter dirodnika. Stan energetyczny tego dirodnika, mam na myśli, czy dirodnik pozostaje głównie w stanie singletowym, czy tripletowym pozostaje otwarte. Co ciekawe, cząsteczka zawiera dwa fragmenty allenowe. Są one dość naprężone, gdyż odpowiedni kąt walencyjny wynosi 134.1° zamiast 180°. Obecność dwóch niesparowanych elektronów sugeruje możliwość di- lub oligomeryzacji cząsteczki.
Manualna zmiana położenia atomów wodoru przy rodnikowych atomach węgla z pozycji endo-endo na położenie exo-exo pozwoliła na utworzenie nowego modelu, który został poddany optymalizacji. Obliczenia pokazały, że nowa cząsteczka zawiera dwa fragmenty allenowe w układzie tricyklicznym (1r,2m,5m,8r)-tricyklo[5.1.0.02,8]octa-2,3,5,6-tetraene.
Wykazanie, czy zmiana położenia atomów wodoru jest łatwa wymagałaby przeprowadzenia obliczeń bariery aktywacji takiego przemieszczenia. To zadanie jest do wykonania. Przeprowadzono dodatkowo rozbudowę dirodnika. Rezultaty tej pracy są pokazane w punkcie 4.2.1. i 4.2.2.
Rozbudowa modelu H do trzech warstw i każdej następnej nieparzystej liczby tych pierścieni (m = 1,3,5…13) powoduje, że pary skrzydłowych atomów wodoru znajdują się na prostych prostopadłych (schemat 4.3.):
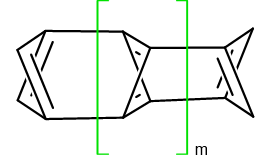
Optymalizacja rozbudowanych struktur daje nowe związki, które albo zachowują pierwotny szkielet, albo ulegają przegrupowaniu. Co ciekawe większa liczba modeli H zachowuje szkielet wyjściowy w porównaniu do szeregu modeli C. Tabela 4.2. pokazuje wyniki obliczeń analogicznie do tabeli 4.1 (niebieski – retencja szkieletu, czerwony – przegrupowanie):
Tabela będzie uzupełniona
Dla modelu długołańcuchowego (m = 14) wyniki optymalizacji nie były jednoznaczne. W jednym wypadku nastąpiła retencja szkieletu a w drugim przegrupowanie (rysunek 4.2.):
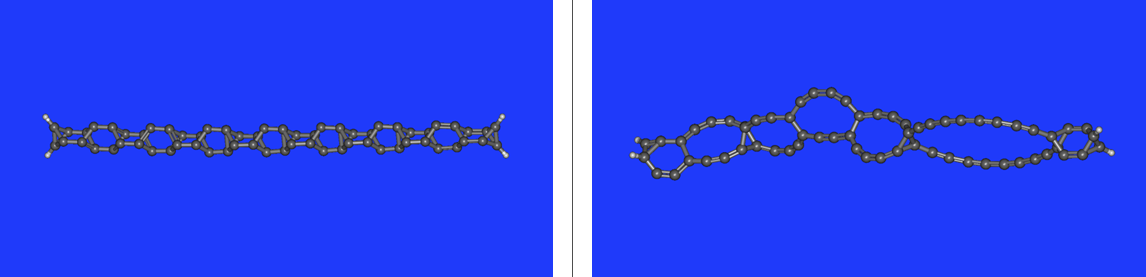
Wprawdzie w pierwszym wypadku uzyskano retencję szkieletu, ale obliczenie częstości na poziomie DFT dało kilka wartości ujemnych. Może to oznaczać, że procedury optymalizacyjne programu Orca w tym konkretnym wypadku nie zdołały przeskoczyć lokalnych maksimów energetycznych podczas obliczeń. Spowodowało to zachowanie szkieletu. Jednakże, ujemne wartości własne hessianu sygnalizują, że cząsteczka powinna mieć inną, optymalniejszą energetycznie, strukturę. Potwierdza to drugi wynik z przegrupowaniem o energii znacznie niższej niż model z retencją struktury. Obie serie obliczeń wykonano na dwóch nieznacznie różnych zbiorach danych optymalizowanych wstępnie za pomocą MM w programie jmol. Zapewne niewielkie różnice położenia atomów w tych dwóch zbiorach xyz spowodowały powstanie dramatycznie różnych struktur wynikowych. To zagadnienie pozostaje również otwarte.
4.2.1. Oligomeryzacja modelu H (m = 0)
Optymalizacja podstawowego modelu H spowodowała jego przegrupowanie i powstanie dirodnika. Sugerowało to możliwość jego oligomeryzacji z utworzeniem nowych cząsteczek. Wykonano takie obliczenia dla pochodnych cyklicznych: dimeru, trimeru i tetrameru (poniżej wszystkie atomy wodoru mają kolor różowy):
O ile struktury dimeru i trimeru były zgodne z pierwotną koncepcją, co oznacza, że w tych wypadkach sytuacja geometryczna spowodowała wytworzenie fragmentów allenowych to optymalna struktura tetrameru stanowi pewną niespodziankę. Bliskość czterech gałęzi allenowych modelu spowodowała, że powstały dodatkowe pierścienie czteroczłonowe kosztem sprzężenia wiązań podwójnych (allenowych).
4.2.2. Odwodornienie oligomerów modelu H
Jedną z motywacji w niniejszej pracy była konstrukcja modeli o jak najmniejszej liczbie atomów wodoru w cząsteczkach węglowodorów. Już w tym miejscu mogę powiedzieć, że celem finalnym było stworzenie nowych odmian alotropowych tego pierwiastka poprzez usunięcie (o ile to było możliwe) wszystkich atomów wodoru z wykreowanych węglowodorów. Aby zbliżyć się do tego celu, w powyżej opisanych oligomerach modelu H zostały usunięte wicynalne atomy wodoru znajdujące się przy tetraedrycznych atomach węgla z jego dimeru, trimeru, tetrameru oraz pentameru (nie badałem wersji uwodornionej pentameru bo jego struktura była po prostu nieładna) w taki sposób, aby powstały wiązania podwójne. Dla porządku pokazano strukturę odwodornionego dimeru. Należy jednak powiedzieć, że hessian dla tego modelu posiada jedną ujemną wartość własną. Oznacza to, że powinien istnieć inny izomer o niższej energii niż pokazany. Tego problemu nie było z odwodornionym trimerem, tetramerem oraz pentamerem. Tam wszystkie częstości obliczone w hessianie były dodatnie.
Należy zwrócić uwagę na różnicę strukturalną pomiędzy tetramerem a jego pochodną odwodornioną. Wynika ona z tego, że cząsteczki odwodornione były tworzone po optymalizacji za pomocą MM a nie po optymalizacji na wyższych poziomach.
5. Podsumowanie
W części pierwszej omówiono systematycznie rozbudowę modeli węglowodorów opartą na tricyklo[3.1.1.12,4]octane oraz jego nienasyconych pochodnych. Do obliczeń kwantowo-chemicznych użyto modeli o maksymalnej liczbie wiązań podwójnych w projektowanych cząsteczkach. Wyniki obliczeń są pokazane powyżej. W zależności od liczby jednostek strukturalnych wyjściowe modele zachowują szkielet wyjściowy lub ulegają przegrupowaniom. Nie jest jasna przyczyna, dla której zachodzi retencja lub transformacja szkieletów. Należy zauważyć, że w wypadku przegrupowań tworzą się dość przypadkowe struktury. Ich cechą charakterystyczną jest obecność długich łańcuchów sprzężonych polienów. Ta ostatnia obserwacja stała się źródłem pomysłów do budowy innych nieznanych dotychczas cząsteczek.